Scientists have created a new method to structure peptides, which they say will be cheaper and make the process of using stapled peptides in drug discovery much more widely available.
The method developed by the nine-strong University of Leeds team, and exploiting synthetic chemistry developed at University College London, is more versatile, cheaper, completely reversible and results in peptides whose pharmacokinetic properties, such as solubility could be easily tuned, according to Professor Andrew Wilson, who led the study.
The approach could allow skilled scientists to constrain a peptide, deliver it to a particular cell and then unconstrain it, preventing it from leaving the cell again.
Making peptides more useful
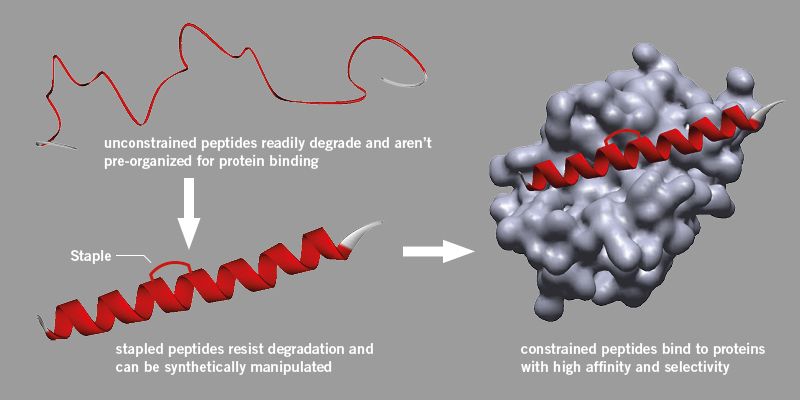
Scientists and drug manufacturers generally regard peptides with scepticism, believing they are too unreliable to be used as drugs because they are easily degraded in cells and are difficult to manipulate.
However research initiated at Harvard University found a method to introduce a brace which could be attached to a peptide, strengthening its active structure and making it more stable and less likely to degrade.
This process, known as ‘stapling’ uses a specific synthetic method and has now become a recognised approach in peptide drug discovery, even resulting in new start-up companies. Stapled peptides do not break down, bind better to proteins and have improved pharmacokinetic properties such as cell uptake.
Protein-protein interactions
A widespread change in perspective on peptides has arisen due to the need for the pharmaceutical industry to develop drugs that act on protein-protein interactions, in contrast to the traditional ‘lock-and-key’ mode of recognition exploited by most current drugs. Small molecules that act on protein-protein interactions must cover a larger less convex surface.
Professor Wilson, whose team works on this problem, said: “Scientists have had limited success in developing traditional small molecules that act on protein-protein interactions, and developing peptides in this way is an attractive alternative.”
He added: “The much greater levels of contact between the peptide and protein can be exploited to achieve higher affinity and selectivity, so we have become excited by this alternative approach to stapling.”
Naturally-occuring amino acids
The University of Leeds team has taken the principle of peptide stapling to a new level. The method is based on the reaction of two thiols in a peptide sequence with a dibromomaleimide reagent.
When the two thiols are correctly placed in the peptide sequence the reaction results in the structure becoming locked in a specific and bioactive helical shape which is more stable.
The method works on peptide sequences containing the natural and common amino acids cysteine or homocysteine. This will permit access to suitable peptides via routine chemical and biological methods.
Professor Wilson, from the University of Leeds’ School of Chemistry and Astbury Centre for Structural Molecular Biology said: “Our approach to protein stapling is more flexible, potentially cheaper, and should be easier to use in chemical biology and drug discovery.
The full research paper Double Quick, Double Click Reversible Peptide ‘Stapling’ is published in the Royal Society of Chemistry’s Chemical Science journal.
Dr Michael Webb, Associate Professor of Chemical Biology at the University of Leeds, said: “We’ve developed a new method to rapidly introduce a functional constraint into two model peptides which enhances a range of biophysical and biochemical properties. Unlike other methods it can also be reversed.
“This approach can be carried out using naturally available amino acids, potentially bringing peptide stapling to a larger community.
“The ability to reverse the constraint may enable delivery of peptide-based reagents into cell where they can be unconstrained and be less easy to transport back out again.”
Further information
Journalists with further questions should email the University of Leeds press office via pressoffice@leeds.ac.uk